
ERCC4

| |
| PDB 1z00 | |
| Identificadores | |
| Símbolo | ERCC4 |
| Símbolos alt. | ERCC11, FANCQ, RAD1, XPF, XFEPS, excision repair cross-complementation group 4, ERCC excision repair 4, endonuclease catalytic subunit |
| Entrez | 2072 |
| RefSeq | NP_005227 |
| UniProt | Q92889 |
| Outros datos | |
| Locus | Cr. 16 16p13.12(13.92 – 13.95 Mb) |
A ERCC4 é unha proteína tamén chamada endonuclease de reparación do ADN XPF, que nos humanos está codificaa no xene ERCC4 do cromosoma 16. Xunto coa proteína ERCC1, ERCC4 forma o complexo encimático ERCC1-XPF, que participa na reparación e recombinación do ADN.[1][2]
O encima nuclease ERCC1-XPF corta estruturas específicas do ADN. Moitos aspectos destes dous produtos xénicos describiranse conxuntamente porque actúan xuntos durante a reparacion do ADN. A actividade da nuclease ERCC1-XPF é esencial na vía de reparación por escisión de nucleótidos do ADN. A nuclease ERCC1-XPF tamén funciona en vías de reparación de roturas de dobre febra do ADN, e na reparación de danos por formación de "enlaces cruzados" que ligan anormalmente as dúas febras do ADN.
As células con lesións incapacitantes no xene ERCC4 son máis sensibles do normal a determinados axentes que danan o ADN, como a radiación ultravioleta e compostos químicos que causan a formación de enlaces cruzados entre as febras do ADN. Os ratos modificados por enxeñaría xenética con mutacións que inhabilitan o xene ERCC4 teñen tamén defectos na reparación do ADN, así como cambios fisiolóxicos inducidos polo estrés metabólico, que orixinan un envellecemento prematuro.[3] A deleción completa de ERCC4 orixina ratos non viables, pero en hmanos non se encontraron individuos cunha deleción completa (homocigota) de ERCC4. Hai casos raros na poboación humana de portadores de mutacións herdadas que impiden o funcionamento de ERCC4. Cando os xenes normais están ausentes, estas mutacións poden orixinar en humanos algunhas síndromes, como o xeroderma pigmentoso, a síndrome de Cockayne ou a anemia de Fanconi.
ERCC4 e ERCC1 son os nomes utilizados para os xenes humanos e Ercc1 e Ercc4 son os nomes dos xenes análogos noutros mamíferos. Xenes similares con funcións semellantes atópanse en todos os organismos eucariotas.
Xene[editar | editar a fonte]
O xene ERCC4 humano pode corrixir os defectos na reparación do ADN en liñas de células mutantes específicas sensibles á luz ultravioleta derivadas de células de ovario de hámster chinés.[4] Illáronse múltiples grupos de complementación independentes de células de ovario do hámster chinés,[5] e este xene restauraba a resistencia á radiación ultravioleta en células do grupo de complementación 4. Facendo referencia a este método de complementación xenética entre especies, o xene foi denominado en inglés "Excision repair cross-complementing 4", de onde proceden as siglas ERCC4[6] A outra denominación, XPF, fai referencia ao xeroderma pigmentoso.
O xene ERCC4 humano codifica a proteína XPF de 916 aminoácidos cunha masa molecular duns 104 000 daltons.
Xenes similares a ERCC4 con funcións equivalentes (ortólogos) encóntranse noutros xenomas eucariotas. Algúns dos xenes ortólogos máis estudados son RAD1 do lévedo de xemación Saccharomyces cerevisiae e rad16+ no lévedo de fisión de Schizosaccharomyces pombe.
Proteína[editar | editar a fonte]

Unha molécula de XPF e outra de ERCC1 únense para formar o heterodímero ERCC1-XPF, que é a forma de nuclease activa do encima. No heterodímero ERCC1–XPF, o ERCC1 media interaccións co ADN e proteína-proteína. O XPF proporciona o sitio activo da endonuclease e está implicado na unión ao ADN e interaccións proteína-proteína adicionais.[4]
A proteína ERCC4/XPF consta de dúas áreas conservadas separadas por unha rexión menos conservada. A área N-terminal ten homoloxía con varios dominios conservados de ADN helicases que pertencen á superfamilia II, aínda que XPF non é unha ADN helicase.[7] A rexión C-terminal de XPF inclúe os residuos do sitio activo para a actividade de nuclease.[8] (Figura 1).
A maior parte da proteína ERCC1 está relacionada a nivel de secuencia co C-terminal da proteína XPF,[9] pero carece dos residuos do dominio de nuclease. Hai un dominio de unión ao ADN "hélice-forquita-hélice" no C-terminal de cada proteína.
Pola súa secuencia primaria e similitude estrutural, a nuclease ERCC1-XPF é membro dunha ampla familia de ADN nucleases específicas de estrutura compostas por dúas subunidades, entre as cales está, por exemplo, a nuclease MUS81-EME1.
Nuclease específica de estrutura[editar | editar a fonte]

O complexo ERCC1–XPF é unha endonuclease específica de estrutura. O ERCC1-XPF só corta o esqueleto fosfodiéster do ADN especificamente nas unións entre o ADN de dobre febra e de febra simple. Introduce un corte no ADN de dobre febra no lado 5′ desa unión, a uns dous nucleótidos de distancia.[10] (Figura 2). Esta especificidade de estrutura foi inicialmente demostrada para RAD10-RAD1, os ortólogos nos lévedos de ERCC1 e XPF.[11]
Os motivos hidrófobos hélice–forquita–hélice nas rexións C-terminais de ERCC1 e XPF interaccionan promocionando a dimerización das dúas proteínas.[12] Non hai actividade catalítica en ausencia de dimerización. Aínda que o dominio catalítico está incluído en XPF, e ERCC1 é cataliticamente inactivo, o ERCC1 é indispensable para a actividade do complexo.
Propuxéronse varios modelos para explicar a unión de ERCC1–XPF ao DNA, baseados en estruturas parciais de fragmentos de proteínas relevantes a resolución atómica.[12] A unión ao ADN mediada polos dominios hélice-forquita-hélice de ERCC1 e dominios XPF posiciona o heterodímero nas zonas de unión entre o ADN de dobre febra e de febra simple.
Reparación por escisión de nucleótidos[editar | editar a fonte]
Durante a reparación por escisión de nucleótidos, varios complexos proteicos cooperan para recoñecer o ADN danado e separan localmente a hélice do ADN durante unha curta distancia a ambos os lados do sitio do dano no ADN. A nuclease ERCC1–XPF fai unha incisión na febra de ADN danado no lado 5′ da lesión.[10] Durante a reparación por escisión de nucleótidos, a proteína ERCC1 interacciona coa proteína XPA para coordinar a unión entre o ADN e a proteína.
Reparación de roturas de dobre febra[editar | editar a fonte]
As células de mamíferos con ERCC1–XPF mutantes son moderadamente máis sensibles que as células normais a axentes (como as radiacións ionizantes) que causan roturas de dobre febra no ADN.[13][14] As vías particulares da reparación por recombinación homóloga e por unión de extremos non homólogos dependen da función de ERCC1-XPF.[15][16] A actividade relevante de ERCC1–XPF para ambos os tipos de reparación de roturas de dobre febra é a súa capacidade de eliminar as colas de febra simple 3' non homólogas dos extremos do ADN antes de volver unilas. Esta actividade é necesaria durante unha subvía de annealing de febra simple da recombinación homóloga. O podado da cola de febra simple 3' é tamén necesaria nunha subvía con mecanismo distinto de unión de extremos non homólogos, dependente das proteínas Ku.[13] A integración homóloga do ADN, unha importante técnica para a manipulación xenética, é dependente da función de ERCC1-XPF na célula hóspede.[17]
Reparación de enlaces cruzados entre febras do ADN[editar | editar a fonte]
As células de mamíferos que portan mutacións en ERCC1 ou XPF son sensibles especificamente a axentes que causan a formación de enlaces cruzados entre as febras do ADN.[18] Os enlaces cruzados entre febras do ADN bloquean a progresión da replicación do ADN, e as estruturas que quedan bloqueadas nas forcadas de replicación do ADN proporcionan substratos para a clivaxe feita por ERCC1-XPF.[19][20] As incisións poden facerse a ambos os lados dos enlaces cruzados nunha febra de ADN para desfacer os enlaces cruzados e iniciar a reparación. Alternativamente, pode facerse unha rotura de dobre febra no ADN preto de enlaces cruzados entre febras, e a subseguinte reparación de recombinación homóloga pode implicar a acción da ERCC1-XPF. Aínda que non é a única nuclease implicada, o ERCC1–XPF é necesario para a reparación de enlaces cruzados entre febras durante varias fases do ciclo celular.[21][22]
Importancia clínica[editar | editar a fonte]
Xeroderma pigmentoso[editar | editar a fonte]
Algúns individuos coa rara síndrome hereditaria xeroderma pigmentoso teñen mutacións en ERCC4. Estes pacientes son clasificados como grupo de complementación F do xeroderma pigmentoso (XP-F). As características de diagnóstico do xeroderma pigmentoso son: pel escamosa seca, pigmentación anormal da pel en áreas expostas ao sol e grave fotosensibilidade, acompañada por unha multiplicación por 1000 do risco de desenvolver cancros de pel inducidos pola radiación ultravioleta.[1]
Síndrome de Cockayne[editar | editar a fonte]
A maioría dos pacientes XP-F mostran síntomas moderados de xeroderma pimentoso, pero uns poucos mostran os síntomas adicionais da síndrome de Cockayne.[23] Os pacientes de síndrome de Cockayne presentan fotosensibilidade e desenvolven síntomas e defectos neurolóxicos.[1][3]
As mutacións no xene ERCC4 poden orixinar a rara síndrome XF-E.[24] Estes pacientes teñen características de xeroderma pigmentoso e de síndrome de Cokayne, así como síntomas adicionais neurolóxicos, hepatobiliares, musculoesqueléticos e hematopoéticos.
Anemia de Fanconi[editar | editar a fonte]
Varios pacientes humanos con síntomas de anemia de Fanconi teñen mutacións que a causan no xene ERCC4. A anemia de Fanconi é unha doenza complexa, principalmente con síntomas hematopoéticos. Unha distinción característica da anemia de Fanconi é a hipersensibilidade a axentes que causan enlaces cruzados entre as febras do ADN. Os pacientes de anemia de Fanconi con mutacións en ERCC4 foron clasificados como pertencentes ao grupo de complementación P da anemia de Fanconi (FANCP).[23][25]
ERCC4 (XPF) no colon normal[editar | editar a fonte]
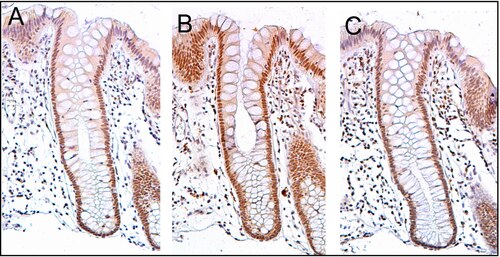
O ERCC4 (XPF) exprésase normalmente a alto nivel nos núcleos celulares na superficie interna do colon (ver imaxe, panel C). A superficie interna do colon está tapizada por un epitelio columnar simple con invaxinacións. As invaxinacións denomínanse glándulas intestinais ou criptas do colon. As criptas do colon teñen forma de tubos de ensaio microscópicos de paredes grosas cun oco central ao longo do tubo (o lume da cripta). As criptas teñen unhas 75 a 110 células de longo. A reparación do ADN, que implica unha alta expresión das proteínas ERCC4 (XPF), PMS2 e ERCC1, parece ser moi activa nas criptas no epitelio do colon normal non neoplástico.
As células orixínanse na base das criptas e migran cara a arriba ao longo do eixe da cripta antes de acabar desprendéndose no lume do colon días despois.[27] Hai 5 ou 6 células nai na base das criptas.[27] Hai uns 10 millóns de criptas ao longo da superficie interna dun colon humano medio.[26] Se as células nai na base da cripta expresan ERCC4 (XPF), xeralmente todos os miles de células da cripta expresan tamén ERCC4 (XPF). Isto vén indicado pola cor marrón que se ve por inmunomarcaxe de ERCC4 (XPF) en case todas as células da cripta no panel C da imaxe deste corte. Unha expresión similar de PMS2 e ERCC1 ocorre nas miles de células de cada cripta de colon normal.
O corte histolóxico da imaxe que se mostra aquí foi tamén contratinguido con hematoxilina para que se tinga o ADN dos núcleos de cor gris azulado. Os núcleos das células na lámina propia, que son células que están debaixo e rodeadas de criptas epiteliais, mostran intensamente a cor gris azulada que lle dá a hematoxilina e presentan pouca expresión de PMS2, ERCC1 ou ERCC4 (XPF). Ademais, as células na parte máis superior das criptas tinguidas para PMS2 (panel A) ou ERCC4 (XPF) (panel C) teñen baixos niveis destas proteínas reparadoras do ADN, polo que estas células mostran tamén a tinguidura gris azulada do ADN.[26]
Deficiencia de ERCC4 (XPF) no epitelio do colon adxacente a ou dentro de cancros[editar | editar a fonte]

O ERCC4 (XPF) é deficiente nun 55% dos cancros de colon, e nun 40% das criptas de colon nun epitelio nos 10 cm adxacentes aos cancros (nos defectos de campo a partir dos cales probablemente se orixina o cancro).[26] Cando ERCC4 (XPF) se reduce nas criptas do colon nun defecto de campo, está máis a miúdo asociado tamén con expresión reducida dos encimas da reparación do ADN de ERCC1 e PMS2, como se ilustra na imaxe desta sección. As deficiencias en ERCC1 (XPF) no epitelio de colon parecen deberse á represión epixenética.[26] Unha deficiencia de ERCC4 (XPF) orixinaría unha redución da reparación dos danos no ADN. Como indicaron Harper e Elledge,[28] os defectos na capacidade de responder axeitadamente e reparar os danos no ADN subxacen en moitas formas de cancro. A redución epixenética frecuente en ERCC4 (XPF) en defectos de campo que rodean os cancros de colon e tamén noutros cancros (xunto con reducións epixenéticas en ERCC1 e PMS2) indica que tales reducións poden a miúdo xogar un papel central na progresión ao cancro de colon.
Aínda que as reducións epixenéticas na expresión de ERCC4 (XPF) son frecuentes en cancros de colon humanos, as mutacións en ERCC4 (XPF) son raros en humanos.[29] Porén, unha mutación no ERCC4 (XPF) causa que os pacientes sexan proclives a ter cancro de pel.[29] Un polimorfismo herdado no ERCC4 (XPF) parece ser importante tamén no cancro de mama.[30] Estas infrecuentes alteracións mutacionais subliñan o probable papel da deficiencia de ERCC4 (XPF) na progresión do cancro.
Notas[editar | editar a fonte]
- ↑ 1,0 1,1 1,2 Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006). DNA Repair and Mutagenesis. ASM Press. ISBN 1555813194.
- ↑ "Entrez Gene: ERCC4 excision repair cross-complementing rodent repair deficiency, complementation group 4".
- ↑ 3,0 3,1 Gregg SQ, Robinson AR, Niedernhofer LJ (Jul 2011). "Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease". DNA Repair 10 (7): 781–91. PMC 3139823. PMID 21612988. doi:10.1016/j.dnarep.2011.04.026.
- ↑ 4,0 4,1 Westerveld A, Hoeijmakers JH, van Duin M, de Wit J, Odijk H, Pastink A, Wood RD, Bootsma D (1984). "Molecular cloning of a human DNA repair gene". Nature 310 (5976): 425–9. PMID 6462228. doi:10.1038/310425a0.
- ↑ Busch D, Greiner C, Lewis K, Ford R, Adair G, Thompson L (Sep 1989). "Summary of complementation groups of UV-sensitive CHO cell mutants isolated by large-scale screening". Mutagenesis 4 (5): 349–54. PMID 2687628. doi:10.1093/mutage/4.5.349.
- ↑ Brookman KW, Lamerdin JE, Thelen MP, Hwang M, Reardon JT, Sancar A, Zhou ZQ, Walter CA, Parris CN, Thompson LH (Nov 1996). "ERCC4 (XPF) encodes a human nucleotide excision repair protein with eukaryotic recombination homologs". Molecular and Cellular Biology 16 (11): 6553–62. PMC 231657. PMID 8887684. doi:10.1128/mcb.16.11.6553.
- ↑ Sgouros J, Gaillard PH, Wood RD (Mar 1999). "A relationship between a DNA-repair/recombination nuclease family and archaeal helicases". Trends in Biochemical Sciences 24 (3): 95–7. PMID 10203755. doi:10.1016/s0968-0004(99)01355-9.
- ↑ Enzlin JH, Schärer OD (Apr 2002). "The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif". The EMBO Journal 21 (8): 2045–53. PMC 125967. PMID 11953324. doi:10.1093/emboj/21.8.2045.
- ↑ Gaillard PH, Wood RD (Feb 2001). "Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair". Nucleic Acids Research 29 (4): 872–9. PMC 29621. PMID 11160918. doi:10.1093/nar/29.4.872.
- ↑ 10,0 10,1 Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD (Sep 1996). "Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease". Cell 86 (5): 811–22. PMID 8797827. doi:10.1016/s0092-8674(00)80155-5.
- ↑ Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC (Sep 1994). "Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease". Science 265 (5181): 2082–5. PMID 8091230. doi:10.1126/science.8091230.
- ↑ 12,0 12,1 McNeil EM, Melton DW (Nov 2012). "DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy". Nucleic Acids Research 40 (20): 9990–10004. PMC 3488251. PMID 22941649. doi:10.1093/nar/gks818.
- ↑ 13,0 13,1 Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ (Aug 2008). "ERCC1-XPF endonuclease facilitates DNA double-strand break repair". Molecular and Cellular Biology 28 (16): 5082–92. PMC 2519706. PMID 18541667. doi:10.1128/MCB.00293-08.
- ↑ Wood RD, Burki HJ, Hughes M, Poley A (Feb 1983). "Radiation-induced lethality and mutation in a repair-deficient CHO cell line". International Journal of Radiation Biology and Related Studies in Physics, Chemistry, and Medicine 43 (2): 207–13. PMID 6600735. doi:10.1080/09553008314550241.
- ↑ Al-Minawi AZ, Saleh-Gohari N, Helleday T (Jan 2008). "The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells". Nucleic Acids Research 36 (1): 1–9. PMC 2248766. PMID 17962301. doi:10.1093/nar/gkm888.
- ↑ Sargent RG, Rolig RL, Kilburn AE, Adair GM, Wilson JH, Nairn RS (Nov 1997). "Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1". Proceedings of the National Academy of Sciences of the United States of America 94 (24): 13122–7. PMC 24273. PMID 9371810. doi:10.1073/pnas.94.24.13122.
- ↑ Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R (Nov 2001). "The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells". The EMBO Journal 20 (22): 6540–9. PMC 125716. PMID 11707424. doi:10.1093/emboj/20.22.6540.
- ↑ Wood RD (Jul 2010). "Mammalian nucleotide excision repair proteins and interstrand crosslink repair". Environmental and Molecular Mutagenesis 51 (6): 520–6. PMC 3017513. PMID 20658645. doi:10.1002/em.20569.
- ↑ Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P (May 2014). "XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4". Molecular Cell 54 (3): 460–71. PMC 5067070. PMID 24726325. doi:10.1016/j.molcel.2014.03.015.
- ↑ Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD (Aug 2000). "Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease". The Journal of Biological Chemistry 275 (34): 26632–6. PMID 10882712. doi:10.1074/jbc.C000337200.
- ↑ Clauson C, Schärer OD, Niedernhofer L (Oct 2013). "Advances in understanding the complex mechanisms of DNA interstrand cross-link repair". Cold Spring Harbor Perspectives in Biology 5 (10): a012732. PMC 4123742. PMID 24086043. doi:10.1101/cshperspect.a012732.
- ↑ Rahn JJ, Adair GM, Nairn RS (Jul 2010). "Multiple roles of ERCC1-XPF in mammalian interstrand crosslink repair". Environmental and Molecular Mutagenesis 51 (6): 567–81. PMID 20658648. doi:10.1002/em.20583.
- ↑ 23,0 23,1 Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li TS, Yoshiura K, Utani A, Hirano A, Yamashita S, Greenblatt D, Nardo T, Stefanini M, McGibbon D, Sarkany R, Fassihi H, Takahashi Y, Nagayama Y, Mitsutake N, Lehmann AR, Ogi T (May 2013). "Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia". American Journal of Human Genetics 92 (5): 807–19. PMC 3644632. PMID 23623389. doi:10.1016/j.ajhg.2013.04.007.
- ↑ Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH (Dec 2006). "A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis". Nature 444 (7122): 1038–43. PMID 17183314. doi:10.1038/nature05456.
- ↑ Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillón J, Ramírez MJ, Pujol R, Casado JA, Baños R, Rio P, Knies K, Zúñiga S, Benítez J, Bueren JA, Jaspers NG, Schärer OD, de Winter JP, Schindler D, Surrallés J (May 2013). "Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia". American Journal of Human Genetics 92 (5): 800–6. PMC 3644630. PMID 23623386. doi:10.1016/j.ajhg.2013.04.002.
- ↑ 26,0 26,1 26,2 26,3 26,4 26,5 26,6 Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, Stern S, Oatman N, Banerjee B, Bernstein C (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr 3 (1): 3. PMC 3351028. PMID 22494821. doi:10.1186/2041-9414-3-3.
- ↑ 27,0 27,1 Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, Simons BD, Jansen M, Graham TA (2014). "Quantification of crypt and stem cell evolution in the normal and neoplastic human colon". Cell Rep 8 (4): 940–7. PMC 4471679. PMID 25127143. doi:10.1016/j.celrep.2014.07.019.
- ↑ Harper JW, Elledge SJ (2007). "The DNA damage response: ten years after". Mol. Cell 28 (5): 739–45. PMID 18082599. doi:10.1016/j.molcel.2007.11.015.
- ↑ 29,0 29,1 Gregg SQ, Robinson AR, Niedernhofer LJ (2011). "Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease". DNA Repair (Amst.) 10 (7): 781–91. PMC 3139823. PMID 21612988. doi:10.1016/j.dnarep.2011.04.026.
- ↑ Lee E, Levine EA, Franco VI, Allen GO, Gong F, Zhang Y, Hu JJ (2014). "Combined genetic and nutritional risk models of triple negative breast cancer". Nutr Cancer 66 (6): 955–63. PMID 25023197. doi:10.1080/01635581.2014.932397.
Véxase tamén[editar | editar a fonte]
Ligazóns externas[editar | editar a fonte]
- Este artigo contén textos actualizados por un experto externo na wikipedia inglesa, que actualizou o correspondente artigo da wikipedia e publicou un artigo académico de "revisión" revisado por pares na revista "Gene", cuxa referencia é: Mandira Manandhar, Karen S Boulware (12 June 2015). "The ERCC1 and ERCC4 (XPF) genes and gene products". Gene. 569 (2): 153–161. doi:10.1016/J.GENE.2015.06.026. PMC 4536074. PMID 26074087.
Galería PDB | |
|---|---|
|


